
Ein Gendefekt beim Barth-Syndrom trifft vor allem die Mitochondrien. Barth-Syndrom-Patienten leiden vor allem unter angeborener Herzmuskelschwäche.
Eine Mutation des Gens „tafazzin“ ist Auslöser für das Barth-Syndrom (BTHS). Die Mutation wird X-chromosomal rezessiv vererbt– Frauen sind nicht betroffen, nur Männer erkranken am Barth-Syndrom. Bald nach der Geburt oder in den ersten Lebensjahren treten die ersten Symptome auf: Herzmuskelschwäche, Neutropenie, allgemeine Myopathie und Wachstumsstörungen.
In 14 Prozent der Fälle präsentiert sich die Hermuskelschwäche dermaßen schwerwiegend, dass eine Herztransplantation notwenig wird. Grundsätzlich lassen sich die Symptome der Krankheit heutzutage aber gut behandeln, die Lebenserwartung der Barth-Syndrom Patienten ist dank der medizinischen Fortschritte deutlich gestiegen.
Das Barth-Syndrom – eine schwere Erbkrankheit
Vor etwa 20 Jahren wurde die genetische Ursache für das Barth-Syndrom entdeckt. Der spezielle Gendefekt führt zu dieser seltenen, aber schweren Erbkrankheit. Betroffene leiden wie erwähnt an verschiedenen Symptomen, besonders schwerwiegend ist eine ausgeprägte Kardiomyopathie (Herzmuskelschwäche). Nicht klar ist, warum sich der Gendefekt vor allem auf Herzmuskelzellen auswirkt.
Forscher der Universitätsmedizin Göttingen (UMG) sind unlängst der molekularen Ursache für diese Besonderheit nachgegangen und entdeckten, dass die Veränderung, die Mutation des Gens zu entscheidenden Veränderungen in den zellulären Energiekraftwerken, den Mitochondrien, führt. Speziell Herzmuskelzellen sind betroffen. Die Störungen beeinträchtigen die Leistung der Mitochondrien, wodurch diese die Herzzellen nur noch beeinträchtigt mit Energie versorgen können.
Die Ergebnisse im Detail
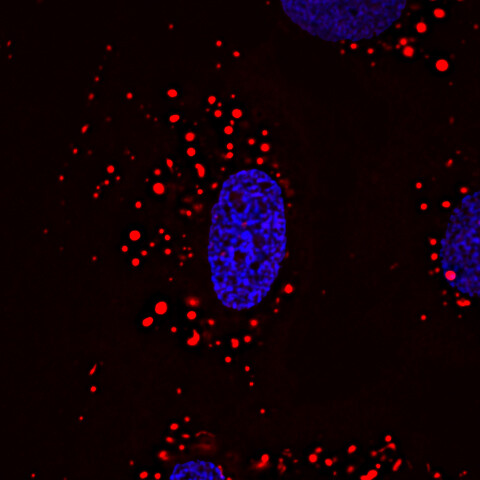
Der Gendefekt bei Patienten mit Barth-Syndrom trifft vor allem die Mitochondrien. Er führt dazu, dass Zellen das mitochondriale Lipid Cardiolipin nicht mehr richtig bilden können. Durch diesen Mangel an reifem Cardiolipin verlieren die Mitochondrien ihre ursprünglich netzwerkartige Form und präsentieren sich mit einer punktförmigen Ausprägung haben (siehe Abbildung). Cardiolipin findet sich vor allem in der inneren Hüllmembran der Mitochondrien und beeinflusst die Anordnung der Proteinkomplexe in der Membran. Sie sorgen somit entscheidend dafür, dass die Mitochondrien richtig arbeiten, um die Zelle mit Energie beliefern zu können.
Warum genau beeinflusst ein Mangel an Cardiolipin die Leistung der Mitochondrien?
Warum führt die Veränderung von Cardiolipin vor allem zu Herzmuskelschwäche? Ein genauerer Blick auf das reife Cardiolipin brachte die Forscher einen entscheidenden Schritt weiter: Seine Zusammensetzung spielt eine wichtige Rolle. Cardiolipin kommt in verschiedenen Geweben in leicht veränderten Formen vor. Um die Form herzustellen, die im Herzen dominiert, ist genau jenes Gen nötig, das bei Barth-Syndrom mutiert ist.
In Untersuchungen von im Labor hergestellten Herzzellen von Barth-Syndrom-Patienten und in Herzen vonBarth-Syndrom-kranken Mäusen konnten die Forscher bemerkenswerte Veränderungen feststellen. In den BTHS-Zellen waren deutlich weniger Superkomplexe in der Mitochondrienmembran. Die Mitochondrien produzierten zudem viel weniger Energie. Offenbar beeinflusst Cardiolipin die Verknüpfung der einzelnen Komplexe zu Superkomplexen, meinen die Wissenschaftler. Passend zu dieser veränderten Situation fanden die Forscher in Barth-Syndrom-Zellen auch mehr freie Radikale als in gesunden Zellen.
Die Forscher beobachteten außerdem, dass einer der Proteinkomplexe, die in den Mitochondrien die Elektronen transportieren, in den Herz-Mitochondrien in ungewöhnlich geringen Mengen vorhanden war. Von jenem Proteinkomplex, dem sogenannten „Komplex II“, war bereits bekannt, dass für seinen Einbau in die Mitochondrienmembran Cardiolipin nötig ist. Außerdem ist Komplex II wichtig, um Fettsäuren für die Energieproduktion nutzen zu können. Das Herz deckt einen Großteil seines Energiebedarfs durch Fettsäuren. Ohne Komplex II hat das Herz also ein Energieproblem.
Ansatzpunkte für Behandlungsstrategien
Mit ihren Erkenntnissen ist den Forschern ein wichtiger Schritt zum Verständnis des Barth-Syndroms gelungen. Daraus ergeben sich auch Ansatzpunkte für eine Behandlungsstrategie. Weniger Komplex II bedeutet eine schlechtere Fettsäureverwertung, weniger Superkomplexe und eine schlechtere Energieproduktion. Mehr freie Radikale verursachen den Zellen zusätzlichen Stress. Dieses Wissen bietet neue Ansatzpunkte, um in Zukunft möglicherweise neue Therapien für BTHS-Patienten zu entwickeln. Es liefert außerdem neue grundsätzliche Erkenntnisse für das Verständnis der Herzmuskelschwäche.
Literatur:
Bertero E, Kutschka I, Maack C, Dudek J. Cardiolipin remodeling in Barth syndrome and other hereditary cardiomyopathies. Biochim Biophys Acta Mol Basis Dis. 2020;1866(8):165803. doi:10.1016/j.bbadis.2020.165803
Dudek J, Cheng IF, Chowdhury A, Wozny K, Balleininger M, Reinhold R, Grunau S, Callegari S, Toischer K, Wanders RJ, Hasenfuß G, Brügger B, Guan K, Rehling P. Dudek J, Cheng IF, Chowdhury A, et al. Cardiac-specific succinate dehydrogenase deficiency in Barth syndrome. EMBO Mol Med. 2016;8(2):139-154. doi:10.15252/emmm.201505644
Ferreira C, Pierre G, Thompson R, Vernon H. Barth Syndrome. In: Adam MP, Ardinger HH, Pagon RA, et al., eds. GeneReviews®. Seattle (WA): University of Washington, Seattle; October 9, 2014.